Plot FastQC data
qc_plot(qc, modules = "all")
# S3 method for qctable
print(x, ...)Arguments
- qc
An object of class qc_read or a path to the sample zipped fastqc result file.
- modules
Character vector containing the names of fastqc modules for which you want to import the data. Default is all. Allowed values include one or the combination of:
"Summary",
"Basic Statistics",
"Per base sequence quality",
"Per sequence quality scores",
"Per base sequence content",
"Per sequence GC content",
"Per base N content",
"Sequence Length Distribution",
"Sequence Duplication Levels",
"Overrepresented sequences",
"Adapter Content",
"Kmer Content"
Partial match of module names allowed. For example, you can use modules = "GC content", instead of the full names modules = "Per sequence GC content".
- x
an object of class qctable.
- ...
other arguments.
Value
Returns a list of ggplots containing the plot for specified modules..
Examples
# Demo file
qc.file <- system.file("fastqc_results", "S1_fastqc.zip", package = "fastqcr")
qc.file
#> [1] "/private/var/folders/xm/8p6yj4bj6s57n4v_51714lwm0000gp/T/RtmpT6jSz8/temp_libpatha9b37e9f6eab/fastqcr/fastqc_results/S1_fastqc.zip"
# Read all modules
qc <- qc_read(qc.file)
#> Reading: /private/var/folders/xm/8p6yj4bj6s57n4v_51714lwm0000gp/T/RtmpT6jSz8/temp_libpatha9b37e9f6eab/fastqcr/fastqc_results/S1_fastqc.zip
#> Warning: Missing column names filled in: 'X1' [1]
#> Warning: Missing column names filled in: 'X1' [1]
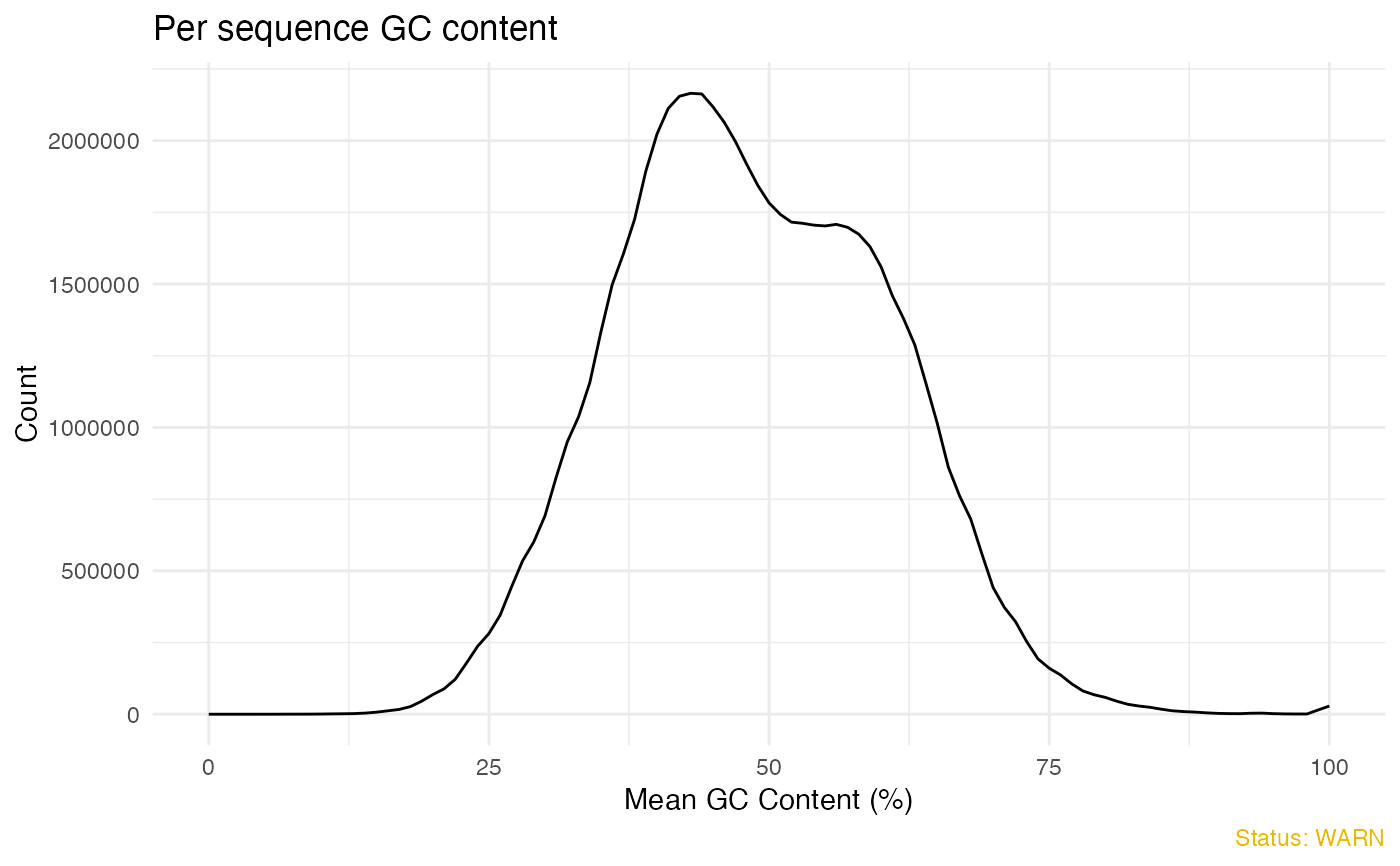
# Plot per sequence GC content
qc_plot(qc, "Per sequence GC content")
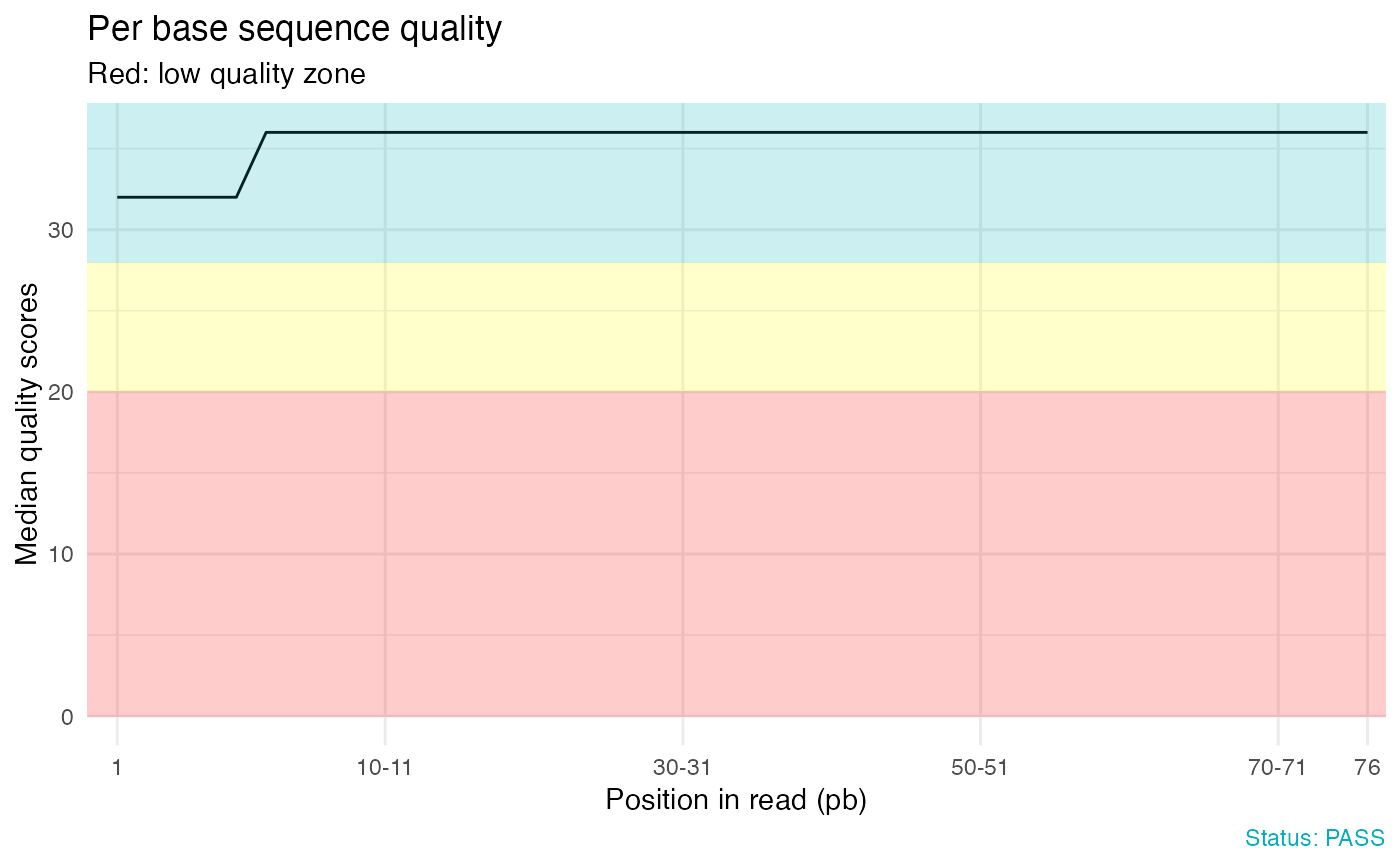
 # Per base sequence quality
qc_plot(qc, "Per base sequence quality")
# Per base sequence quality
qc_plot(qc, "Per base sequence quality")
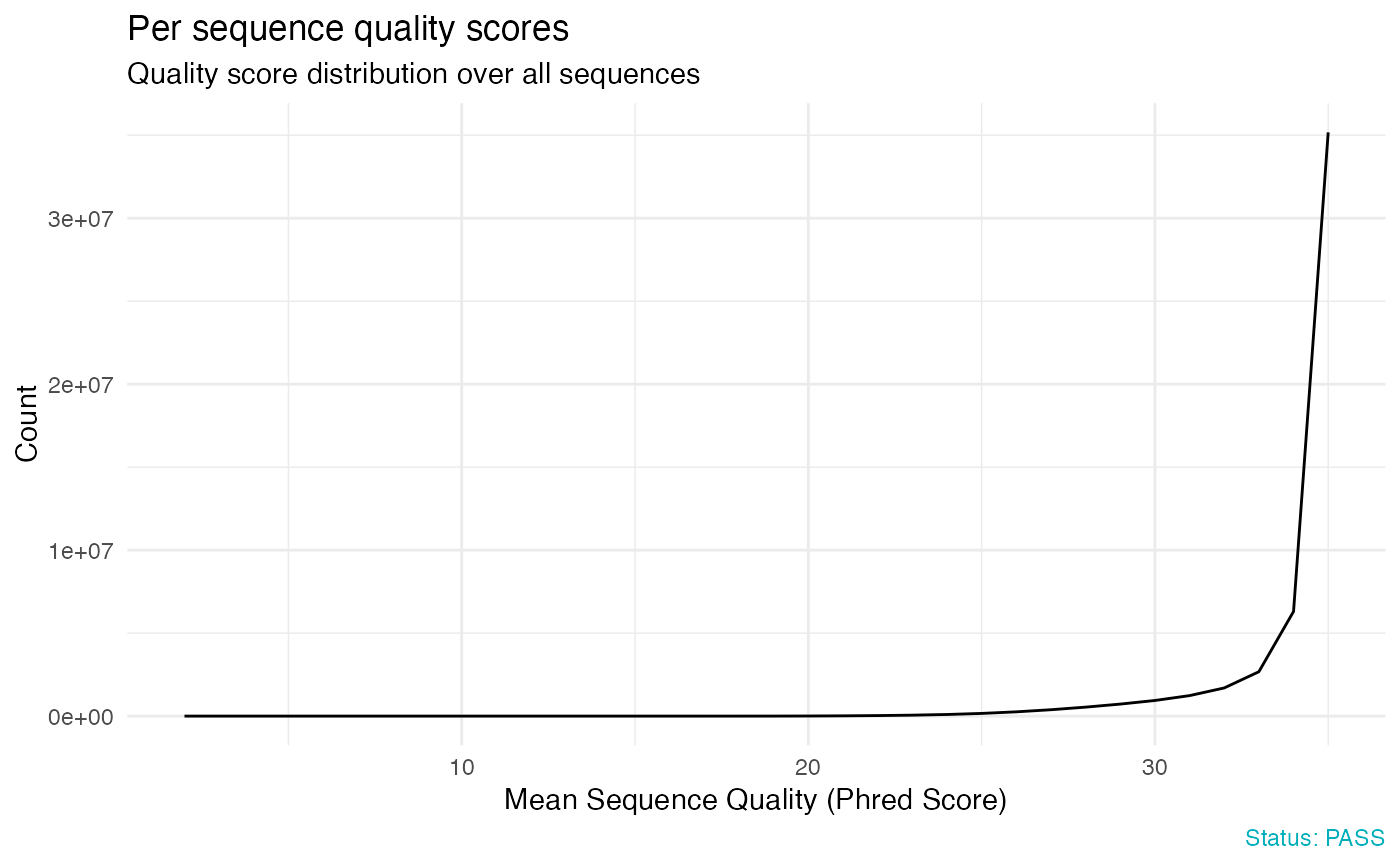
 # Per sequence quality scores
qc_plot(qc, "Per sequence quality scores")
# Per sequence quality scores
qc_plot(qc, "Per sequence quality scores")
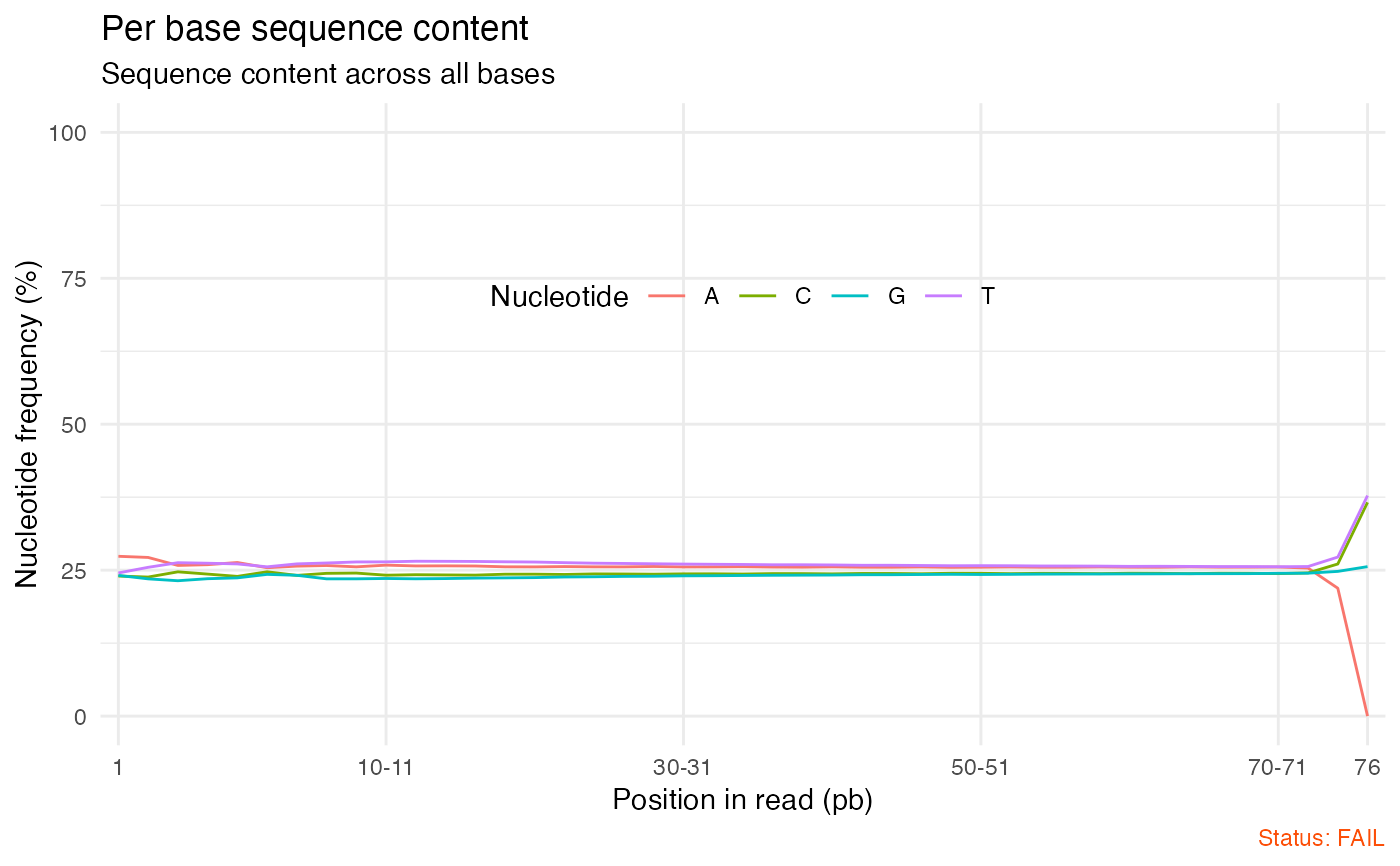
 # Per base sequence content
qc_plot(qc, "Per base sequence content")
# Per base sequence content
qc_plot(qc, "Per base sequence content")
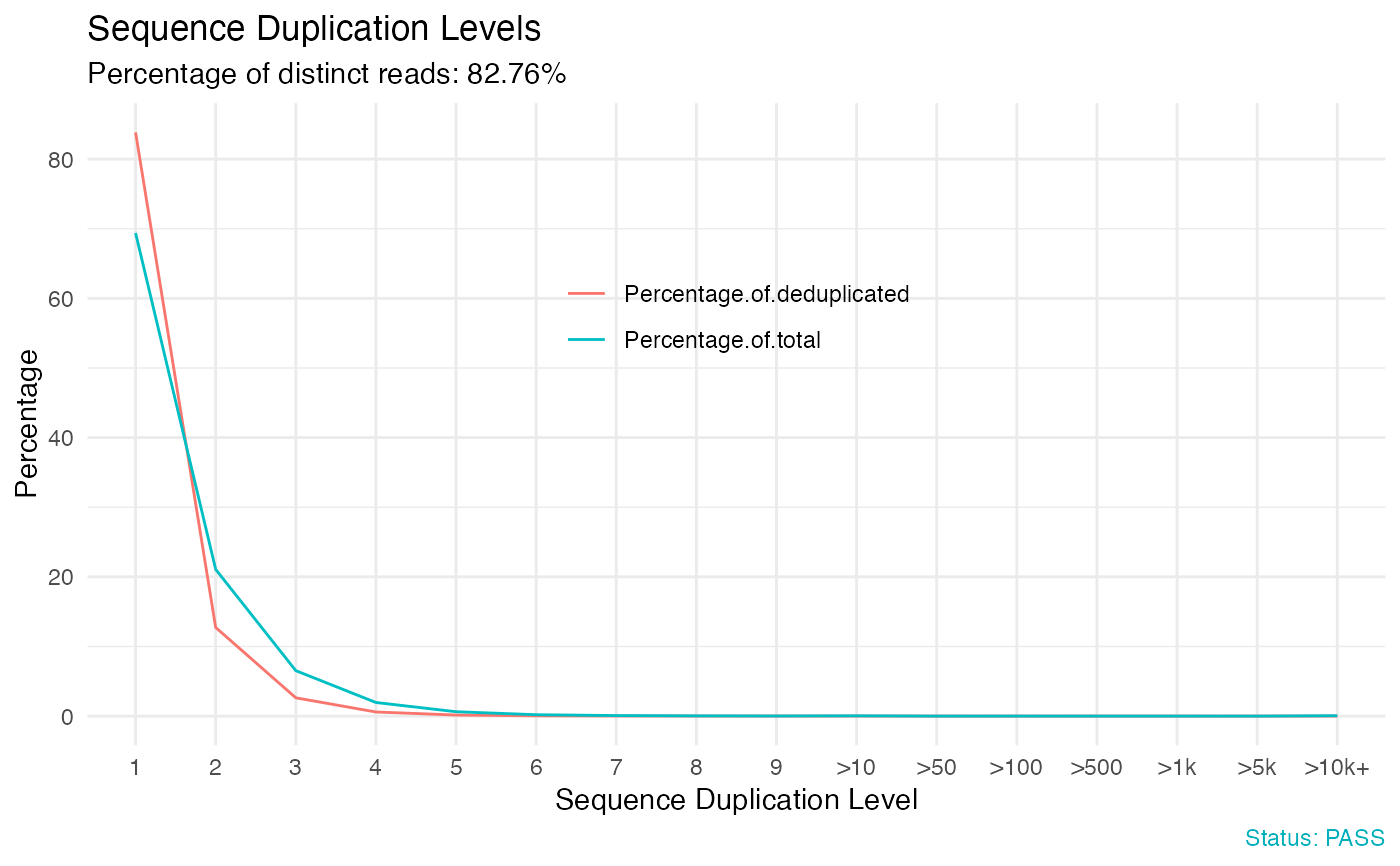
 # Sequence duplication levels
qc_plot(qc, "Sequence duplication levels")
# Sequence duplication levels
qc_plot(qc, "Sequence duplication levels")